BioStudio SDK ¶
The biocolabsdk package empowers users to handle data from all applications within the BioTuring ecosystem.
Installation ¶
To install biocolabsdk, python version 3.7 or greater is required.
pip3 install biocolabsdk -U
Settings ¶
For initial use, you should configure your tokens in the ‘User Settings’ page by following these step:
Step 1: Click on your avatar.
Step 2: Select ‘User Settings’.
Step 3: Configure the following tokens:
- Bioflex API Key: a public Bioflex token used to access data from the BioTuring public database.
To obtain a Bioflex token, you can directly submit a request by following the instruction provided under the ‘Bioflex API Key’ box. - Talk2Data API Key: a token that allows access to the Talk2Data Ecosystem.
- BBrowserX Private Server: a domain that enables access BBrowserX on your private server.
- BBrowserX Private API Key: an API token to access BBrowserX on your private server.
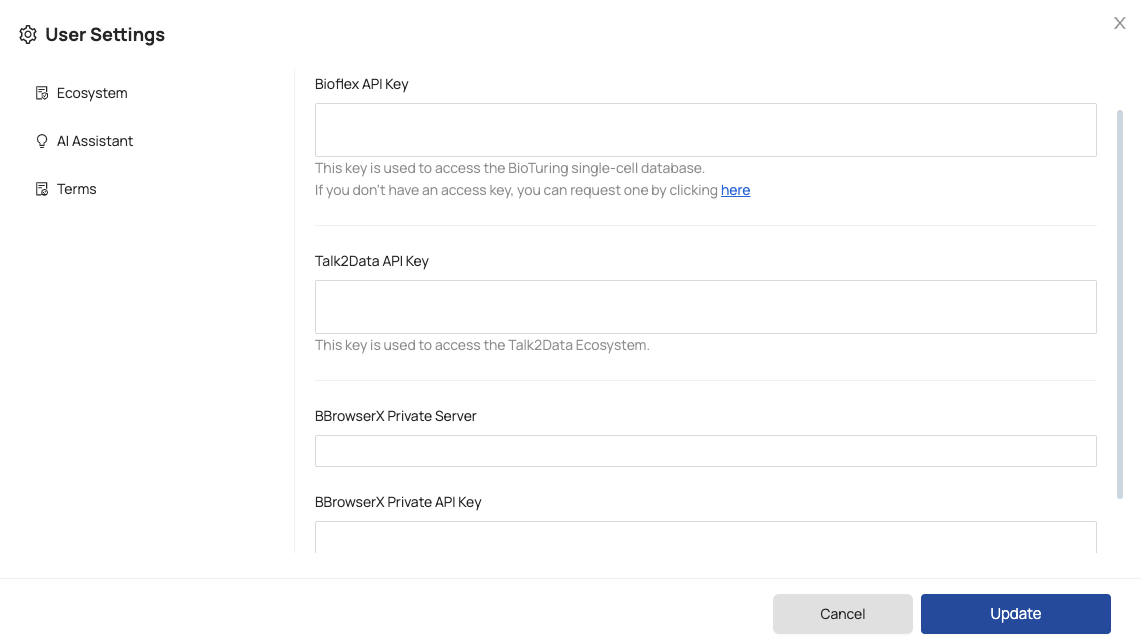
Once you have entered the required information, click on the ‘Update’ button to save the settings.
Submitting data from BioStudio to BBrowserX ¶
If you want to visualize your data after performing analysis in BioStudio, you have the option to submit the data directly from BioStudio to BBrowserX.
Step by step, the following example illustrates how to submit a dataset:
- Step 1: Import required packages
from biocolabsdk import EConnector
from bioturing_connector.typing import InputMatrixType
from bioturing_connector.typing import Species
from bioturing_connector.typing import StudyType
- Step 2: Initiate connection to BBrowserX
Once you have configured all your tokens in the ‘User Settings’ page, you can simply call the method as follows:
connector = EConnector()
To change the domain and its tokens, you have two options. First, you can update the ‘User Settings’ page. Alternatively, you can directly define the private host and token while initializing the connection.
connector = EConnector(
private_host="https://yourcompany/t2d_index_tool/",
private_token="<input your token here>"
)
- Step 3: Test connection to BBrowserX
connector.get_bbrowserx().test_connection()
Example output:
Connecting to host at https://yourcompany/t2d_index_tool/api/v1/test_connection
Connection successful
- Step 4: Get user groups available for your token
user_groups = connector.get_bbrowserx().get_user_groups()
print(user_groups)
Example output:
[{'id': 'all_members', 'name': 'All members'}, {'id': 'personal', 'name': 'Personal workspace'}]
- Step 5: Submit data to BBrowserX
Submit h5ad (scanpy object)
# Submitting the scanpy object from s3:
batch_info = [{
'matrix': 's3_path/GSE128223_1.h5ad',
}, {
'matrix': 's3_path/GSE128223_2.h5ad',
}]
connector.get_bbrowserx().submit_study_from_s3(
group_id='personal',
batch_info=batch_info,
study_id='GSE128223',
name='This is my first study',
authors=['Huy Nguyen'],
species=Species.HUMAN.value,
input_matrix_type=InputMatrixType.RAW.value,
study_type=StudyType.H5AD.value
)
# Submitting the scanpy object from local machine:
batch_info = [{
'matrix': 'local_path/GSE128223_1.h5ad',
}, {
'matrix': 'local_path/GSE128223_2.h5ad',
}]
connector.get_bbrowserx().submit_study_from_local(
group_id='personal',
batch_info=batch_info,
study_id='GSE128223',
name='This is my first study',
authors=['Huy Nguyen'],
species=Species.HUMAN.value,
input_matrix_type=InputMatrixType.RAW.value,
study_type=StudyType.H5AD.value
)
Example output:
> [2022-10-10 01:03] Waiting in queue
> [2022-10-10 01:03] Downloading GSE128223.h5ad from s3: 262.1 KB / 432.8 MB
> [2022-10-10 01:03] File downloaded
> [2022-10-10 01:03] Reading batch: GSE128223.h5ad
> [2022-10-10 01:03] Preprocessing expression matrix: 19121 cells x 63813 genes
> [2022-10-10 01:03] Filtered: 19121 cells remain
> [2022-10-10 01:03] Start processing study
> [2022-10-10 01:03] Normalizing expression matrix
> [2022-10-10 01:03] Running PCA
> [2022-10-10 01:03] Running kNN
> [2022-10-10 01:03] Running spectral embedding
> [2022-10-10 01:03] Running venice binarizer
> [2022-10-10 01:04] Running t-SNE
> [2022-10-10 01:04] Study was successfully submitted
> [2022-10-10 01:04] DONE !!!
> Study submitted successfully!
In addition to h5ad, you can submmit data with a wige range of data input format such as 10X, H5, RDS, and TSV.
Example input batch_info for 10X format:
batch_info = [{
'name': 'Batch 1',
'matrix': 'path_to/batch_1/matrix.mtx1',
'features': 'path_to/batch_1/features.tsv',
'barcodes': 'path_to/batch_1/barcodes.tsv',
}, {
'name': 'Batch 2',
'matrix': 'path_to/batch_2/matrix.mtx1',
'features': 'path_to/batch_2/features.tsv',
'barcodes': 'path_to/batch_2/barcodes.tsv',
}]
Available parameters for submit_study function:
group_id: str
ID of the group to submit the data to.
study_id: str, default=None
Study ID, if no value is specified, use a random uuidv4 string
name: str, default='To be detailed'
Name of the study.
authors: List[str], default=[]
Authors of the study.
abstract: str, default=''
Abstract of the study.
species: str, default='human'
Species of the study. Can be: **bioturing_connector.typing.Species.HUMAN.value** or **bioturing_connector.typing.Species.MOUSE.value** or
**bioturing_connector.typing.Species.NON_HUMAN_PRIMATE.value**
input_matrix_type: str, default='raw'
If the value of this input is **bioturing_connector.typing.InputMatrixType.NORMALIZED.value**, then the software will use slot 'X' from the scanpy object and does not apply normalization.
If the value of this input is **bioturing_connector.typing.InputMatrixType.RAW.value**,then the software will use slot 'raw.X' from thescanpy object and apply log-normalization.
skip_dimred: bool, default='False'
If the input is a scanpy/seurat object, this parameter can be set to True to skip BioTuring pipeline (PCA/UMAP/t-SNE)
study_type: str, default='h5ad'
The format of the study. Can be: **bioturing_connector.typing.StudyType.MTX_10X.value** or **bioturing_connector.typing.StudyType.H5.value**
or **bioturing_connector.typing.StudyType.H5AD.value** or **bioturing_connector.typing.StudyType.RDS.value** or **bioturing_connector.typing.StudyType.TSV.value**
min_counts: str, default=None
Minimum number of counts required for a cell to pass filtering.
min_genes: int, default=None
Minimum number of genes expressed required for a cell to pass filtering.
max_counts: int, default=None
Maximum number of counts required for a cell to pass filtering.
max_genes: int, default=None
Maximum number of genes expressed required for a cell to pass filtering.
mt_percentage: Union[int, float], default=None
Maximum number of mitochondria genes percentage required for a cell to pass filtering. Ranging from 0 to 100
Importing data from BBrowserX to BioStudio ¶
BioStudio enable users to clone their data from BBrowserX to BioStudio for their own further analyses.
Please follow the instruction bellow for detailed guidance:
Import required packages
import numpy as np
from biocolabsdk import EConnector
from bioturing_connector.typing import InputMatrixType
Initiate connection to BBrowserX
Once you have configured all your tokens in the ‘User Settings’ page, you can simply call the method as follows:
connector = EConnector()
To change the domain and its tokens, you have two options. First, you can update the ‘User Settings’ page. Alternatively, you can directly define the private host and token while initializing the connection.
connector = EConnector(
private_host="https://yourcompany/t2d_index_tool/",
private_token="<input your token here>"
)
Test connection to BBrowserX
connector.get_bbrowserx().test_connection()
Example output:
Connecting to host at https://yourcompany/t2d_index_tool/api/v1/test_connection
Connection successful
Get user groups available for your token
user_groups = connector.get_bbrowserx().get_user_groups()
print(user_groups)
Example output:
[{'id': 'all_members', 'name': 'All members'}, {'id': 'personal', 'name': 'Personal workspace'}]
Get all study information in a group
study_list = connector.get_bbrowserx().get_all_studies_info_in_group(
group_id='personal',
species='human',
)
print(study_list)
Example output:
[{'uuid': 'dec12f167c6d4686b5837a18ddc2628d', 'study_title': 'PBMC 8k', 'study_hash_id': 'pbmc_8k', 'created_by': 'data@bioturing.com'}]
You can now access your desired dataset using the study_id, which corresponds to its uuid.
Get basic information
Get barcodes:
barcodes = np.array(connector.get_bbrowserx().get_barcodes(
study_id='a363c9345859406d90c170aaef61437b',
species='human',
))
barcodes
Example output:
array(['AAACCCAGTCGGCCTA-1', 'AAACCCATCAGATGCT-1', 'AAACGAAAGATTAGCA-1',
..., 'TTTGTTGGTCTTCAAG-1', 'TTTGTTGTCGAAGTGG-1',
'TTTGTTGTCGCATAGT-1', dtype='<U18'])
Get features:
features = np.array(connector.get_bbrowserx().get_features(
study_id='a363c9345859406d90c170aaef61437b',
species='human',
))
features
Example output:
array(['5S_RRNA', '5_8S_RRNA', '7SK', ..., 'AL121908.1', 'AP000527.1',
'AL035681.1', dtype='<U26'])
Get metadata as a pandas dataframe:
metadata = connector.get_bbrowserx().get_metadata(
study_id='a363c9345859406d90c170aaef61437b',
species='human',
)
metadata
Example output:
| Barcodes | AUCell scores - AUCell score | ... | Number of mRNA transcripts | Percentage of mitochondrial genes | |
| 0 | AAACCCAGTCGGCCTA-1 | 0 | ... | 1083 | 0 |
| 1 | AAACCCATCAGATGCT-1 | 0 | ... | 1083 | 0 |
| 2 | AAACGAAAGATTAGCA-1 | 0 | ... | 1083 | 0 |
| ... | ... | ... | ... | ... | ... |
| 5137 | TTTGTTGGTCTTCAAG-1 | 0 | ... | 1083 | 0 |
| 5138 | TTTGTTGTCGAAGTGG-1 | 0 | ... | 1083 | 0 |
| 5139 | TTTGTTGTCGCATAGT-1 | 0 | ... | 1083 | 0 |
Access the expression matrix
Query a gene:
gene_exp = connector.get_bbrowserx().query_genes(
study_id='a363c9345859406d90c170aaef61437b',
species='human',
gene_names=['CD3D', 'CD8A'],
unit=InputMatrixType.RAW.value,
)
gene_exp
Example output:
<5140x2 sparse matrix of type '<class 'numpy.float32'>'
with 30 stored elements in Compressed Sparse Column format>
Extract the whole expression matrix:
matrix = connector.get_bbrowserx().query_genes(
study_id='a363c9345859406d90c170aaef61437b',
species='human',
gene_names=[],
unit=InputMatrixType.RAW.value,
)
matrix
Example output:
<5140x63751 sparse matrix of type '<class 'numpy.float32'>'
with 359800 stored elements in Compressed Sparse Column format>
Available parameters for query_genes function:
unit: str
If the value of this input is **bioturing_connector.typing.InputMatrixType.NORMALIZED.value**, then the return value will be normalized data.
If the value of this input is **bioturing_connector.typing.InputMatrixType.RAW.value**,then the return value will raw count.
gene_names: List[str], default=[]
If the value array is empty, the return value will be the whole matrix
Access PCA/t-TSNE/UMAP result
List all available embedding results:
emb_list = connector.get_bbrowserx().list_all_custom_embeddings(
study_id='a363c9345859406d90c170aaef61437b',
species='human',
)
emb_list
Example output:
[
{'embedding_id': '74f54f3e96d6453fabc82503cfec3210', 'embedding_name': 'PCA (no batch corrected)'},
{'embedding_id': 'd7a2b82fd2084701851732412d2987de', 'embedding_name': 'UMAP (n_neighbors=30)'},
{'embedding_id': '52e119b665e04648aab567d51f99eeac', 'embedding_name': 'PCA (Harmony)'},
]
Retreive a embedding result:
pca = connector.get_bbrowserx().retrieve_custom_embedding(
study_id='a363c9345859406d90c170aaef61437b',
species='human',
embedding_id= '74f54f3e96d6453fabc82503cfec3210'
)
pca
Example output:
array([[ -3.9508643, -5.0433464 , 11.238977 , ..., 1.3024445 ,
0.09807181 , 0.35462874],
[ 2.695733, -7.363346 , 5.159652 , ..., -2.3540812 ,
-0.7354203 , -0.85309833],
[ 4.099449, -4.446068 , 8.5328045 , ..., 0.23611104 ,
0.49040127 , -1.2935507],
...,
[ 11.718925, -1.6622849 , 3.1962256 , ..., 1.7455299 ,
1.0112557 , -1.5500767],
[ 9.51906, -4.9529567 , -4.8548193 , ..., 1.9500092 ,
-0.6865985 , -0.87388307],
[ -10.006671, 34.502968 , -7.6862097 , ..., 1.6238164 ,
-0.9188056 , 0.5598281]], dtype=float32)
Accessing data from Talk2Data ecosystem ¶
In addition to BBrowserX, the biocolabsdk also supports users to access data from other applications within the Talk2Data ecosystem.
To get started, you need to initiate a connection to the Talk2Data public server.
from biocolabsdk import EConnector
connector = EConnector(
bioflex_token="<input your token here>"
)
connector.get_talk2data().test_connection()
If you have configured all your tokens in the ‘User Settings’ page, you can simply call the method as follows:
from biocolabsdk import EConnector
connector = EConnector()
connector.get_talk2data().test_connection()
Example output:
Connecting to host at https://talk2data.bioturing.com/t2d_index_tool/api/v1/test_connection
Connection successful
You now can utilize all the features in a similar way to accessing a BBrowserX private server.
Accessing BioTuring databse via Bioflex ¶
During your analysis in the BioStudio workspace, you have the capability to clone datasets from the BioTuring database to support your research.
Follow the instruction bellow to clone a dataset:
Import required packages
import bioflex
from biocolabsdk import EConnector
Create connection using access token
If you have configured all your tokens in the ‘User Settings’ page, you can simply call the method as follows:
connector = EConnector()
To change your token, you have two options. First, you can update the ‘User Settings’ page. Alternatively, you can directly define the token while initializing the connection.
connector = EConnector(
bioflex_token="<input your token here>"
)
List available databases
databases = connector.get_bioflex().databases()
databases
Example output:
[DataBase(id="5010c7d573ae4ff2b9691422b99aa2cd",
name="BioTuring database",species="human",version=1),
DataBase(id="5010c7d573ae4ff2b9691422b99aa2cd",
name="BioTuring database",species="human",version=2),
DataBase(id="5010c7d573ae4ff2b9691422b99aa2cd",
name="BioTuring database",species="human",version=3)]
Get database cell types gene expression summary
database = databases[2]
result = database.get_celltypes_expression_summary(['CD3D', 'CD3E'])
print(result['CD3D'][:5])
print(result['CD3E'][:5])
Example output:
[Summary(name="B cell",sum=707108874.0,mean=4192.709686217774,rate=0.03504117106973723,count=168652.0,total=3172495), Summary(name="CD4-positive, alpha-beta T cell",sum=9489987442.0,mean=4657.561967741555,rate=0.5283278751435854,count=2037544.0,total=2751088), Summary(name="CD4-positive, alpha-beta cytotoxic T cell",sum=342799107.0,mean=4684.903951018846,rate=0.5532527824824582,count=73171.0,total=91026), Summary(name="CD8-positive, alpha-beta T cell",sum=8799563254.0,mean=4704.7405575715065,rate=0.5471126656122398,count=1870361.0,total=2452094), Summary(name="CD8-positive, alpha-beta cytotoxic T cell",sum=411976171.0,mean=4748.566944835058,rate=0.5942491575111647,count=86758.0,total=105282)]
[Summary(name="B cell",sum=569738449.0,mean=4098.277566375819,rate=0.028884262036286558,count=139019.0,total=3172495), Summary(name="CD4-positive, alpha-beta T cell",sum=10050349852.0,mean=4702.274442320307,rate=0.5542041025880377,count=2137338.0,total=2751088), Summary(name="CD4-positive, alpha-beta cytotoxic T cell",sum=362243512.0,mean=4758.973068131059,rate=0.5755353254294702,count=76118.0,total=91026), Summary(name="CD8-positive, alpha-beta T cell",sum=9239057247.0,mean=4722.38210576353,rate=0.5722922492023789,count=1956440.0,total=2452094), Summary(name="CD8-positive, alpha-beta cytotoxic T cell",sum=376955768.0,mean=4697.736447247077,rate=0.5496177977478836,count=80242.0,total=105282)]
Create study instance, using study hash ID from BioTuring studies
study = database.get_study('GSE96583_batch2')
study
Example output:
Study(id="GSE96583_batch2",
title="Multiplexed droplet single-cell RNA-sequencing using natural genetic variation (Batch 2)",
reference="https://www.nature.com/articles/nbt.4042")
Take a peek at study metadata
study.metalist
Example output:
[Metadata(id=0,name="sequencing platform - subgroup (standardized)",type="Category"),
Metadata(id=1,name="tissue - subgroup (standardized)",type="Category"),
Metadata(id=2,name="storage technique (standardized)",type="Category"),
Metadata(id=3,name="condition (standardized)",type="Category"),
Metadata(id=4,name="condition - subgroup (standardized)",type="Category"),
Metadata(id=5,name="sequencing platform (standardized)",type="Category"),
Metadata(id=6,name="cell type (standardized)",type="Category"),
Metadata(id=7,name="cell type - subgroup (standardized)",type="Category"),
Metadata(id=8,name="tissue (standardized)",type="Category"),
Metadata(id=9,name="sampling site (standardized)",type="Category"),
Metadata(id=10,name="sampling technique (standardized)",type="Category"),
Metadata(id=11,name="quantification (standardized)",type="Category"),
Metadata(id=12,name="quantification - subgroup (standardized)",type="Category"),
Metadata(id=13,name="Author's cell type",type="Category"),
Metadata(id=14,name="Condition",type="Category"),
Metadata(id=15,name="Louvain clustering",type="Category"),
Metadata(id=16,name="Multiplets",type="Category"),
Metadata(id=17,name="Number of genes",type="Numeric"),
Metadata(id=18,name="Number of mRNA transcripts",type="Numeric"),
Metadata(id=19,name="Percentage of mitochondrial genes",type="Numeric"),
Metadata(id=20,name="Quantification",type="Category"),
Metadata(id=21,name="Sample ID",type="Category"),
Metadata(id=22,name="Sampling site",type="Category"),
Metadata(id=23,name="Sampling technique",type="Category"),
Metadata(id=24,name="Sequencing platform",type="Category"),
Metadata(id=25,name="Stimulation",type="Category"),
Metadata(id=26,name="Storage technique",type="Category"),
Metadata(id=27,name="Tissue",type="Category")]
Fetch a study metadata
Choose a desire metadata field
metadata = study.metalist[4]
metadata
Example output:
Metadata(id=4,name="condition - subgroup (standardized)",type="Category")
Fetch metadata
metadata.fetch()
metadata.values
Example output:
array(['systemic lupus erythematosus', 'systemic lupus erythematosus',
'systemic lupus erythematosus', ..., 'normal', 'normal', 'normal'],
dtype='<U28')
Query genes
exp_mtx = study.query_genes(['CD3D', 'CD3E'], bioflex.UNIT_LOGNORM)
exp_mtx
Example output:
<29065x2 sparse matrix of type '<class 'numpy.float32'>'
with 15492 stored elements in Compressed Sparse Column format>
Get study barcodes
study.barcodes()
Example output:
['GSM2560249_AAACATACCAAGCT-1',
'GSM2560249_AAACATACCCCTAC-1',
...
'GSM2560249_AATTGTGATTCACT-1',
'GSM2560249_AATTGTGATTTCGT-1',
...]
Get study features
study.features()
Example output:
['5S_RRNA',
'5_8S_RRNA',
...
'AC006273',
'AC006277',
...]
Get study full matrix
study.matrix(bioflex.UNIT_LOGNORM)
Example output:
<29065x64642 sparse matrix of type '<class 'numpy.float32'>'
with 17570739 stored elements in Compressed Sparse Column format>
Export Study
study.export_study(bioflex.EXPORT_H5AD)
Example output:
{'download_link': 'https://talk2data.bioturing.com/api/export/6fdaf990b0d14924959b792df5de9f29',
'study_hash_id': 'GSE96583_batch2'}