




Notebooks




Premium


BioTuring

BioTuring

BioTuring

BioTuring
Trends
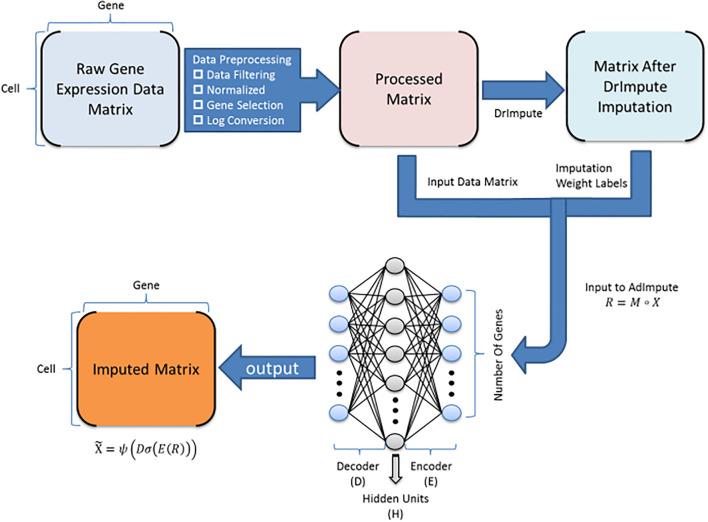
BioTuring
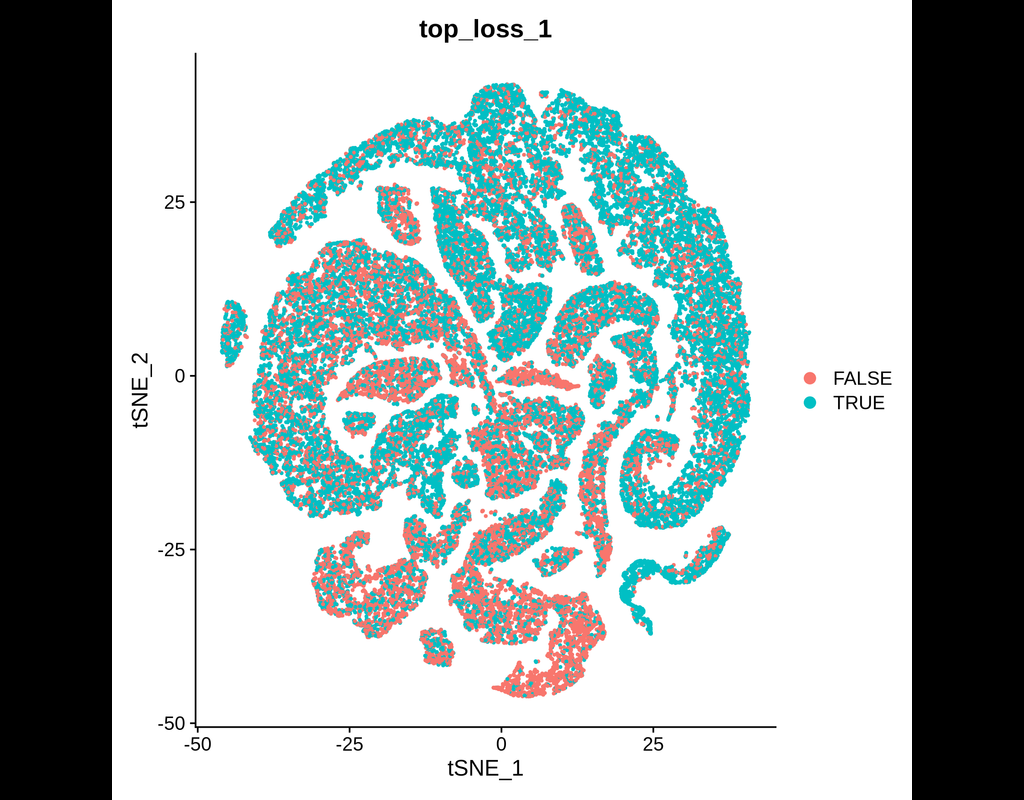
BioTuring
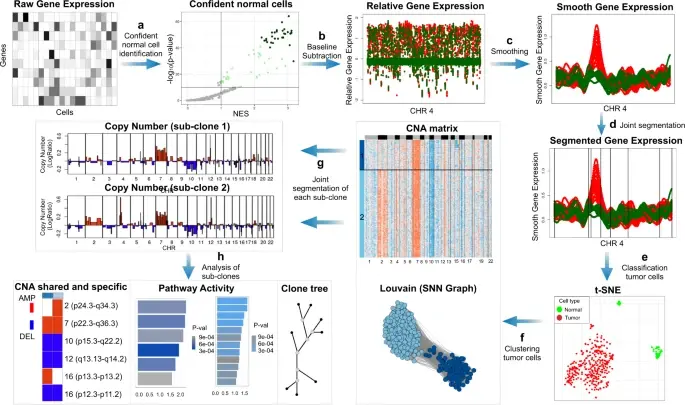
BioTuring
BioTuring

BioTuring

BioTuring

BioTuring
BioTuring

BioTuring
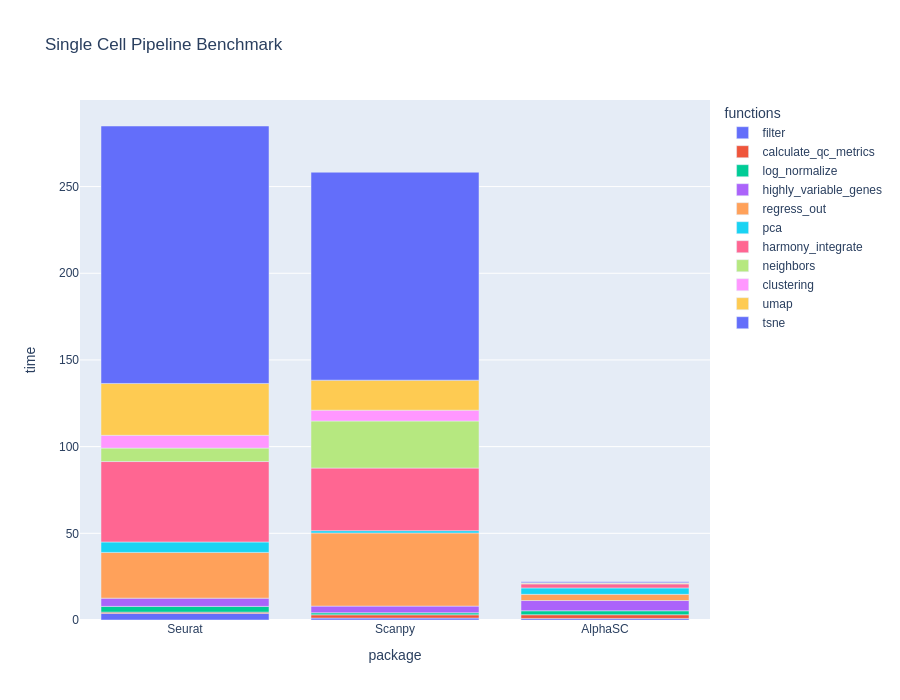
BioTuring

BioTuring
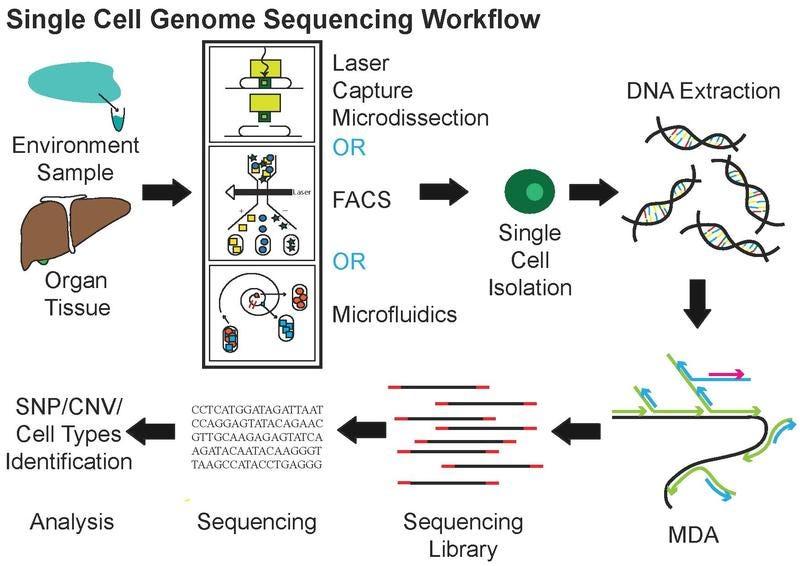
BioTuring

BioTuring
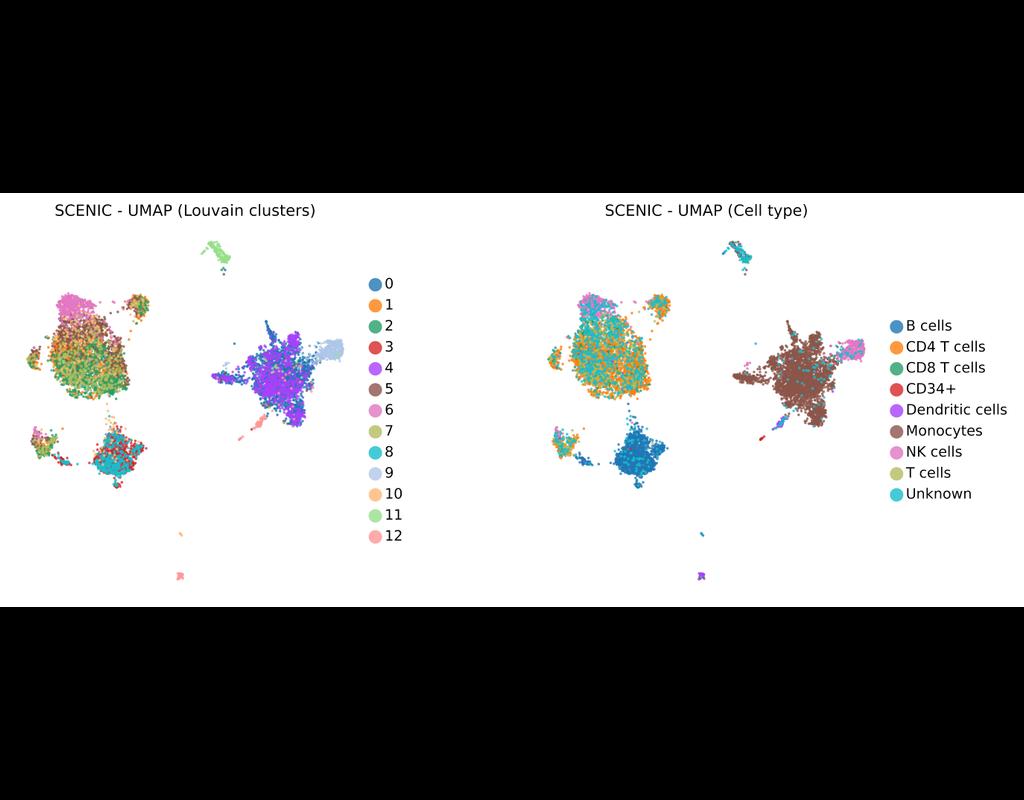
BioTuring
BioTuring
| Notebooks |
|---|